Tamara Schenekar
Zusammenfassung
Für Arterfassungen auf Basis von Umwelt-DNA (environmental DNA, eDNA) wird aus einer Umweltprobe DNA gewonnen und diese z. B.
mittels Metabarcoding-Analyse sequenziert, wodurch mit minimaler Invasivität und sehr zeit- und kosteneffizient Informationen über
die Anwesenheit ganzer Artengemeinschaften generiert werden können. Den vielen Vorteilen eDNA-basierter Erhebungen stehen
allerdings noch einige offene Fragen gegenüber, die vor einer Implementierung dieser Ansätze in standardisierte
Monitoringprogramme geklärt werden müssen, z. B. hinsichtlich der Aussagekraft in Bezug auf Abundanzen oder hinsichtlich der
Dynamik von eDNA in der Umwelt. Nicht nur für Metabarcoding-Analysen sind digitale Sequenzinformationen eine wichtige
Datengrundlage in Naturschutz und Biodiversitätsforschung. Öffentlich zugängliche Sequenzdatenbanken wie GenBank und das Barcode
of Life Data System (BOLD) erlauben der Wissenschaft rasante Fortschritte in vielen Forschungsbereichen. Allerdings steht der
offene Zugang zu generierten Sequenzdaten evtl. im Konflikt mit dem Nagoya-Protokoll, das die faire Nutzung genetischer Ressourcen
und einen gerechten Vorteilsausgleich zwischen den Vertragsstaaten regelt. Hier sind klare und einfache Lösungen erforderlich, um
digitale Sequenzinformationen fair und effizient für Forschung und Biodiversitätsschutz einsetzen zu können.
Umwelt-DNA – Biodiversitätsmonitoring – Barcoding – International Barcode of Life (iBOL) – DNA-Sequenzen – Übereinkommen über die biologische Vielfalt – Nagoya-ProtokollAbstract
For eDNA-based species surveys, DNA is extracted from an environmental sample and sequenced using, for example, metabarcoding
analysis. This allows the generation of information on the presence of entire species assemblages with minimal invasiveness and in
a very time- and cost-efficient manner. The numerous advantages of eDNA-based surveys are counterbalanced by open questions
regarding, e. g., inferences on abundances or eDNA-dynamics in the environment, which need to be clarified before implementing
these approaches in standardised monitoring programmes. Digital sequence information is an important data basis, not only for
metabarcoding analyses, but for nature conservation and biodiversity research in general. Open-access sequence databases such as
GenBank and Barcode of Life Data System (BOLD) allow science to make rapid progress in many fields of research. However, making
generated sequence data public may conflict with the provisions of the Nagoya Protocol on the fair use of genetic resources and
equitable benefit-sharing among signatory states. Clear and simple solutions are needed to use digital sequence information for
research and biodiversity conservation effectively and in a fair manner.
Environmental DNA – Biodiversity monitoring – Barcoding – International Barcode of Life (iBOL) – DNA sequences – Convention on Biological Diversity – Nagoya ProtocolInhalt
1 Einleitung
Das menschengemachte drastische Artensterben stellt eine der größten Herausforderungen unserer Zeit dar. Schätzungen zufolge
sterben Arten derzeit etwa 1.000-fach schneller aus, als es durch natürliche Prozesse im Laufe der Erdgeschichte bisher geschah
( Pimm et al. 2014). Um dem entgegenzuwirken, brauchen wir ein klares Verständnis
dafür, wo gefährdete Organismengruppen vorkommen, um diese dort gezielt schützen zu können. Die Arterfassung in einem bestimmten
Gebiet, ein wichtiger Teil jedes Biodiversitätsmonitorings, stellt daher einen essenziellen Grundpfeiler vieler Naturschutzprojekte
dar. Die Methodik zur Erfassung der Arten variiert zwischen den einzelnen Organismengruppen (
Abb. 1a – c): Die Vogeldiversität wird bspw. meist durch direkte Beobachtung, akustisches Monitoring oder
Nistkastenkontrollen erfasst ( Südbeck 2005). Daten zu Amphibien werden entweder durch
direkte Beobachtung oder durch Abfangen mit Hilfe von Keschern oder Fallen erhoben ( Kupfer,
Schlüpmann 2009), und für das Monitoring von Säugetierarten kommen weltweit großteils indirekte Beobachtungen, etwa
unter Einsatz von Kamerafallen, zur Anwendung. Wasserinsekten, eine wichtige Indikatorgruppe für die Überprüfung der Zielvorgaben der
Europäischen Wasserrahmenrichtlinie (WRRL), werden mittels Kescher in den jeweiligen Gewässern gesammelt und unter dem Mikroskop
morphologisch bestimmt ( Meier et al. 2006). Das Monitoring von Fischen erfolgt für
standardisierte Erhebungen per Elektrobefischung ( Dussling 2009).

Abb. 1: Konventionelle Monitoringmethoden sowie eine eDNA-Beprobung für Biodiversitätserhebungen. a) Elektrobefischung
zur Fischbestandserhebung. b) Vogelmonitoring mit Hilfe von Spektiven. c) Montage einer Kamerafalle zur Überwachung von
Säugetieren. d) Für eine Fließgewässer-eDNA-Beprobung wird ein vordefiniertes Volumen Wasser entnommen. e) Das Wasser wird
durch einen Filter gedrückt, auf dem die DNA-Moleküle haften bleiben. f) Der Filter enthält die aufgefangene eDNA, aber
auch andere Schwebstoffe aus dem Wasser, wodurch er sich verfärbt. eDNA = environmental DNA (Umwelt-DNA).
(Fotos: a) Wolfgang Hauer BAW/IGF, b) Matthias Weissensteiner, c) Jo Taylor, d) – f) Immanuel Karner)
Fig. 1: Conventional biomonitoring methods as well as eDNA sampling for biodiversity assessments. a) Electrofishing for
fish-stock surveys. b) Bird monitoring using spotting scopes. c) Camera trapping for surveying mammals. d) For eDNA sampling
of a riverine system, a predefined volume of water is collected. e) The DNA molecules are captured through filtration. f) The
filter contains the captured eDNA but also other suspended solids from the water, causing it to discolour.
eDNA = environmental DNA.
Diese über Jahrzehnte etablierten und international anerkannten Methoden stoßen jedoch unter gewissen Bedingungen an ihre Grenzen.
So funktionieren Elektrobefischungen in kleinen Gewässern sehr gut, während bei größeren oder trüberen Gewässern die Effizienz stark
abnimmt ( Lyon et al. 2014). Kamerafallen detektieren kleine Säugetiere
deutlich schlechter als große und können von Wildtieren oder Menschen beschädigt oder gestohlen werden ( Glover-Kapfer et al. 2019). Zusätzlich wird mit der zunehmend prekären Situation vieler
Wildtierpopulationen der Ruf lauter, auf invasive Erhebungsmethoden, wie das Fangen per Falle oder Elektrobefischung, zu verzichten,
vor allem bei besonders gefährdeten oder sensiblen Arten.
Aufgrund der Notwendigkeit effizienter und minimal-invasiver Monitoringmethoden und beschleunigt durch damit einhergehende
technologische Entwicklungen hat sich in den letzten 20 Jahren das DNA-basierte Biodiversitätsmonitoring als neuer Ansatz entwickelt,
vor allem jenes mit Hilfe von Umwelt-DNA (environmental DNA, eDNA). Der vorliegende Artikel befasst sich mit dem Einsatz von
eDNA-basierten Ansätzen für Arterfassungen und dem Metabarcoding, bei dem große Mengen digitaler Sequenzinformationen (DSI) erzeugt
werden ( Abschnitt 2), sowie mit der Bedeutung von DSI im Allgemeinen für die
Biodiversitätsforschung ( Abschnitt 3 und
Abschnitt 4), auch abseits von eDNA.
2 eDNA – das neue Schweizer Taschenmesser für Biodiversitätserhebungen?
Der Begriff eDNA beschreibt genetisches Material, das aus Umweltproben gewonnen wird. DNA, also das genetische Erbgut, das in fast
jeder Zelle von Lebewesen enthalten ist, wird von diesen laufend in die Umgebung abgegeben, bspw. durch den Verlust von
Oberflächenzellen wie Hautschuppen bei Tieren oder von Epidermiszellen bei Pflanzen, durch die Abgabe von Pollenstaub, Sporen oder
Samen bei Pflanzen und von Sperma, Eizellen oder Fäkalien bei Tieren ( Taberlet et al.
2012; Abb. 2a, b). Die genaue Definition von eDNA wird noch immer
diskutiert, da bspw. keine Einigkeit darüber besteht, ob der Begriff auch DNA aus gesamten Mikroorganismen wie Bakterien oder
Mikroalgen in Böden oder Biofilmen umfasst oder ob er sich nur auf DNA beziehen soll, die aus Ausscheidungen von Makroorganismen
stammt und bei deren Gewinnung nicht der komplette Zielorganismus aus der Umwelt isoliert wird (
Pawlowski et al. 2020; Rodriguez-Ezpelata et al. 2021). Für den
vorliegenden Beitrag, der sich vor allem auf höhere Vielzeller bezieht, wird letztere Definition verwendet, man spricht hier oft auch
von eDNA im engeren Sinne.
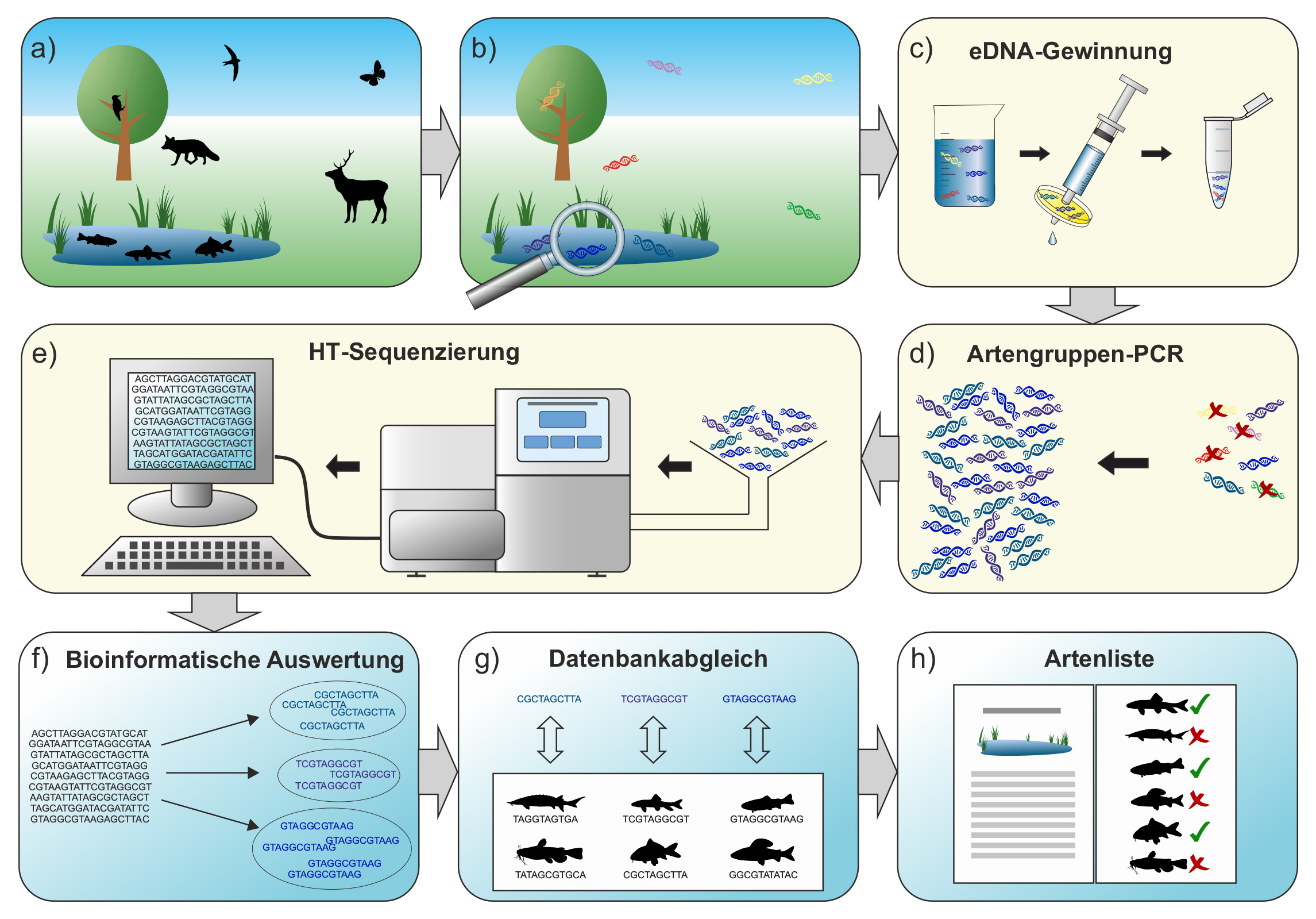
Abb. 2: Workflow einer eDNA-basierten Erhebung am Beispiel einer Metabarcoding-Studie über Fische. a) Tiere bewegen
sich im oder durch das Untersuchungsgebiet, so auch Fische in einem Stillgewässer. b) Diese Tiere hinterlassen ihre DNA in
der Umgebung. c) Eine definierte Wassermenge wird dem Gewässer entnommen. Die eDNA wird durch Filtration konzentriert und
mittels DNA-Extraktion aus dem Filter gewonnen. d) In einer PCR mit fischspezifischen Primern werden nur DNA-Fragmente von
Fischen im eDNA-Extrakt vermehrt. e) Nach einigen Vorbereitungsschritten werden die DNA-Sequenzen der PCR-Produkte mit
Hilfe eines HT-Sequenziergeräts ausgelesen. f) Die Rohsequenzdaten werden in mehreren Schritten (wie Qualitätsfilterungen
und Gruppieren identischer DNA-Sequenzen) bioinformatisch aufbereitet. g) Die DNA-Sequenzen der aufbereiteten Daten werden
mit einer genetischen Referenzdatenbank der potenziell vorkommenden Fische im Gewässer abgeglichen. h) Eine Liste aller
detektierten Arten wird erstellt. eDNA = environmental DNA (Umwelt-DNA); HT = high throughput;
PCR = Polymerase-Kettenreaktion.
Fig. 2: Workflow of an eDNA-based assessment using the example of a metabarcoding study targeting fishes. a) Animals move
in or through the study area, including fish in a still water body. b) These animals leave their DNA in the environment. c) A
predefined volume of water is taken from the water body. The eDNA is concentrated by filtration and DNA extraction is
performed on the filter. d) Through PCR using fish-specific primers, only the DNA fragments of fishes in the eDNA extract are
amplified. e) After some preparatory steps, the DNA sequences of the PCR products are read using an HT sequencer. f) The raw
sequence data are bioinformatically processed in several steps (such as quality filtering and clustering of identical DNA
sequences). g) The DNA sequences of the processed data are compared to a genetic reference database containing the fishes
potentially occurring in the water body. h) A list of species detected is compiled. eDNA = environmental DNA; HT = high
throughput; PCR = polymerase chain reaction.
Je nach Bedingungen im Milieu, in das die DNA abgegeben wird, z. B. im Wasser eines Sees oder in der Umgebungsluft, verweilt eDNA
eine gewisse Zeit in dieser Umgebung, bevor sie zerfällt, abtransportiert oder durch Mikroorganismen abgebaut wird ( Barnes, Turner 2016). Entnimmt man nun eine eDNA-Probe aus der Umwelt und weist in dieser die
DNA bestimmter Tier- oder Pflanzenarten nach, kann man auf die rezente Anwesenheit dieser Arten in der näheren Umgebung schließen. Das
bei Weitem häufigste Medium, aus dem eDNA isoliert wird, ist Wasser ( Beng, Corlett
2020), u. a. weil man eDNA aus Wasser recht einfach durch Filtration auffangen und konzentrieren kann. Hierbei wird ein
definiertes Wasservolumen durch sehr feine Poren filtriert, wobei die DNA-Moleküle vom Filter aufgefangen werden ( Abb.1d – f ; Abb.2c ). Die eDNA-Gewinnung aus
anderen Substraten (wie Sedimenten, Erde oder Luft) unterscheidet sich je nach Medium ( Bohmann
et al. 2014; Deiner et al. 2017). Als Nächstes wird die eDNA durch eine
DNA-Extraktion aus der Probe gewonnen (im Falle filtrierter Wasserproben aus dem Filter). Der darauffolgende Schritt richtet sich nach
der jeweiligen Fragestellung der Studie, wobei es zwei Hauptansätze gibt:
Für Ansatz 1 wird eine artspezifische Polymerase-Kettenreaktion (PCR) am eDNA-Extrakt durchgeführt. Diese PCR amplifiziert
(vermehrt) nur die DNA der Zielart, sofern im eDNA-Extrakt vorhanden. Kommt es nun zu einer Amplifikation der Zielart-DNA, gilt die
Art als detektiert. Dieser Einzelartansatz wird vor allem bei besonders gefährdeten oder sehr versteckt lebenden Arten angewandt. So
kartierten Forscherinnen und Forscher z. B. das Vorkommen von Grottenolmen (Proteus anguinus) im verzweigten Höhlensystem des
dinarischen Karsts mittels artspezifischer PCR ( Vörös et al. 2017), und bereits 2011
nutzten amerikanische Forscherinnen und Forscher diese Methodik, um die Ausbreitung zweier asiatischer Karpfenarten im Kanalsystem um
den Lake Michigan zu belegen und vor einer weiteren Ausbreitung dieser invasiven Arten im See zu warnen. Hierbei erwies sich der
eDNA-Ansatz als viel sensitiver als die konventionelle Elektrobefischung ( Jerde et al.
2011).
Für Ansatz 2 wird meist das so genannte Metabarcoding herangezogen, das für großräumige und viele Arten umfassende
Biodiversitätserhebungen als eine besonders revolutionäre Methodik angesehen wird ( Deiner et al.
2017; Ruppert et al. 2019). Hier wird ebenso eine PCR am eDNA-Extrakt
durchgeführt, allerdings ist diese spezifisch für eine höhere taxonomische Ebene, also eine Gruppe von Arten wie Blütenpflanzen,
Säugetiere oder Insekten. In der PCR wird die DNA aller Arten dieses Zieltaxons, sofern im eDNA-Extrakt vorhanden,
amplifiziert ( Abb. 2d). Im Anschluss werden die DNA-Sequenzen dieser Amplifikate mittels
High-throughput-Sequenzierung (HTS) ausgelesen ( Abb. 2e). Die verarbeiteten Rohsequenzen
werden mit einer genetischen Referenzdatenbank, die die jeweils zugehörigen DNA-Sequenzen der unterschiedlichen Arten des Ziel-Taxons
beinhaltet, abgeglichen ( Abb. 2f, g). Findet man Übereinstimmungen zwischen den
DNA-Sequenzen aus der eDNA-Probe und der Referenzsequenz einer bestimmten Art, gilt diese als detektiert ( Abb. 2h). Im Gegensatz zu Ansatz 1 werden durch HTS DNA-Sequenzen als DSI erzeugt. Diese
liefern nicht nur Informationen über die Präsenz oder Abwesenheit bestimmter Arten, sondern enthalten noch zusätzliche Informationen,
z. B. über die genetischen Distanzen zwischen den detektierten Arten oder die genetische Diversität innerhalb der detektierten Arten.
Daher können sie für eine Vielzahl weiterer Fragestellungen derselben oder aber auch anderer Forschungsgruppen genutzt werden (siehe
Abschnitt 3).
Die HTS hat hierbei in den letzten zwei Jahrzehnten rasante technologische Fortschritte gemacht, so dass es nun möglich ist, große
Datenmengen (bis zu mehrere Milliarden DNA-Sequenzen) in wenigen Stunden und Tagen zu produzieren (
Van Dijk et al. 2014). Des Weiteren ermöglicht die steigende Automatisierung von Prozessen zur Probenvorbereitung für
die HTS einen zunehmend höheren Probendurchsatz in den Laboranalysen ( Buchner et al.
2021). Durch die hohen Durchsätze ist es möglich, für eine große Menge an Probepunkten in relativ kurzer Zeit
Informationen über die Anwesenheiten vieler Arten zu erzeugen, was diese Methode so effizient für Biodiversitätserhebungen macht. Die taxonomische Ebene, also
die Breite der Zielgruppe, kann durch die Auswahl so genannter universeller Primer definiert werden, wobei man hierbei einen
Kompromiss zwischen Universalität (Zielgruppenbreite) und Sensitivität (Fähigkeit, unterschiedliche Arten voneinander unterscheiden zu
können) wählen muss. Stat (2017) beschreibt mit Hilfe weniger Metabarcoding-Marker die
Artenvielfalt in marinen Sedimenten sogar über das gesamte Spektrum der lebendigen Welt, was die immense Universalität dieser Ansätze
belegt.
Im Rahmen der Entwicklung universeller Metabarcoding-Primer für Fische testeten Forscherinnen und Forscher das Verfahren in vier
Becken des zweitgrößten Aquariums der Welt, dem Okinawa Churaumi Aquarium. Hierbei wiesen sie mittels eDNA-Metabarcoding 168 der
180 Fischarten (93,3 %) in den Becken nach, ohne dabei auch nur einen einzigen Fisch fangen zu müssen ( Miya et al. 2015). In Panama extrahierten Biologinnen und Biologen eDNA von Schmeißfliegen
(Calliphoridae), die sich von Tierkadavern, offenen Wunden oder Tierkot ernähren, um DNA-Spuren der Tiere nachzuweisen, von denen sie
Nahrung aufgenommen hatten. Die Ergebnisse der Metabarcoding-Analyse wurden mit jenen von Kamerafallen und Transektzählungen
verglichen. Anhand der eDNA-basierten Ergebnisse konnten 20 Säugetierarten nachgewiesen werden, während Kamerafallen und
Transektzählungen nur 17 bzw. 13 Arten detektierten ( Rodgers et al. 2017). Neueste
Untersuchungen zeigen, dass selbst die Luft ein geeignetes Medium zur Gewinnung von eDNA ist. So gelang es anhand gefilterter Luft aus
Tiergärten, eine Vielzahl der Wirbeltiere, die in und um diese Tiergärten vorkamen, und selbst die DNA von Futtertieren nachzuweisen
( Clare et al. 2022; Lynggaard et al.
2022).
Auf Grund der vielen Vorzüge eDNA-basierter Erhebungen (vgl. Kasten 1) wird der Ruf
lauter, diese in standardisierte Monitoringprogramme bspw. der WRRL oder der Meeresstrategie-Rahmenrichtlinie zu integrieren ( Darling et al. 2017; Hering et al. 2018). Zu
klären bleibt in diesem Zusammenhang z. B., ob man konventionelle Bioindikatoren beibehalten kann oder ob für eDNA-basierte Erhebungen
neue Bioindikatoren und Indizes entwickelt werden müssen ( Pawlowski et al. 2021). Auch
ist die Abschätzung absoluter Abundanzen (Häufigkeiten) von Arten mittels eDNA-basierter Ansätze noch stark umstritten, da sich aus
Sequenzierdaten nur relative Abundanzparameter ableiten lassen ( Rees et al. 2014;
Gloor et al. 2017; Harper et al.
2019), wohingegen absolute Abundanzen ein wichtiger Parameter vieler Monitoringprogramme sind.
Kasten 1: eDNA-basierte Biodiversitätserhebungen – die wichtigsten Stärken und offene Fragen.
Box 1: Key strengths of and open questions about eDNA-based biodiversity assessments.
Vorteile:
● Nichtinvasivität bzw. minimale Invasivität bei der Probennahme: Besonders beim Nachweis von
Wirbeltieren werden diese bzw. deren Lebensraum kaum bis gar nicht gestört. ● Höhere Detektionsraten oder Sensitivität im Vergleich zu vielen konventionellen Methoden, vor allem
bei seltenen oder gefährdeten Arten. ● Universalität der Probennahme: Während sich bei konventionellen Methoden die Felderhebungen bzw.
Probennahmen oft stark zwischen Artengruppen unterscheiden, kann in ein- und derselben eDNA-Probe ein sehr breites
Spektrum an Arten nachgewiesen werden. ● Zeit- und Kosteneffizienz: eDNA-basierte Erhebungen sind teilweise bereits günstiger als konventionelle
Erhebungen. Mit den weiter fallenden Sequenzierkosten wird sich dieser Trend voraussichtlich weiter
fortsetzen. ● Relativ simple Probennahme: Dies erlaubt auch den Einsatz von Nichtspezialistinnen und Nichtspezialisten
oder Citizen Scientists in sehr groß angelegten Monitoringprojekten.
Offene Fragen:
● Es ist ein noch besseres Verständnis der Dynamiken von eDNA in der Umwelt notwendig, also z. B. Antworten
auf die Frage, wie lange eDNA in den jeweiligen Medien verweilt, bevor sie abgebaut oder abtransportiert
wird. ● Noch fehlen klar definierte Methodenstandards, um vergleichbare und robuste Ergebnisse über mehrere Studien
und verschiedene Labore hinweg zu produzieren. ● Aussagen über Abundanzen, also die Individuenanzahl oder Biomasse von Arten, sind noch sehr umstritten, vor
allem beim Metabarcoding. ● eDNA-basierte Erhebungen können derzeit noch keine verlässliche Information über wichtige demographische
Parameter wie Altersstrukturen, Geschlechterverhältnis oder Reproduktionsraten geben. Hier könnte das ganz
neue Feld rund um eRNA neue Erkenntnisse liefern. ● Verfügbarkeit kompletter und qualitativ hochwertiger Referenzsequenzdatenbanken für
Metabarcodinganalyse.
Abkürzungen: DNA = Desoxyribonukleinsäure; eDNA = environmental DNA (Umwelt-DNA); eRNA = environmental RNA (Umwelt-RNA);
RNA = Ribonukleinsäure (ein weiterer Teil des genetischen Materials in Zellen).
Zusammengefasst nach Bohmann et al. (2014);
Barnes, Turner (2016); Deiner et al. (2017); Hering et al. (2018); Pawlowski et al.
(2018); Belle et al. (2019);
Harper et al. (2019); Weigand et al. (2019); Macher et al. (2021).
Ein weiterer wichtiger Punkt für die Implementierung eDNA-basierter Erhebungen ist die Definition von Methodenstandards für
Probennahme, Laboranalysen und Datenauswertung, da nur dadurch miteinander vergleichbare und reproduzierbare Ergebnisse gewährleistet
werden können ( Darling et al. 2017; Pawlowski et al.
2021), und der rigorose Einsatz von Qualitätskontrollen, um z. B. Kontaminationen während der Probenbearbeitung, für die
eDNA-basierte Analysen recht anfällig sind, ausschließen zu können. Schlussendlich ist die Verfügbarkeit möglichst
vollständiger und qualitativ hochwertiger genetischer Referenzdatenbanken eine Grundvoraussetzung für Metabarcoding-basierte
Monitoringprogramme. Denn nur jene Arten, für die genetische Referenzsequenzen vorliegen, lassen sich auch nachweisen. Des Weiteren
hat sich gezeigt, dass sich bei mehreren verfügbaren Referenzsequenzen je Spezies die Identifikationsgenauigkeit deutlich erhöht
( Kolter, Gemeinholzer 2020). Jedoch sind Referenzdatenbanken je nach Tier- oder
Pflanzengruppe bzw. geografischer Region teilweise noch sehr lückenhaft. So errechneten Weigand
et al. (2019), dass für über 80 % der europäischen Fische mindestens eine Referenzsequenz verfügbar ist, dies jedoch auf
weniger als 15 % der europäischen Kieselalgen zutrifft. Neben der Vollständigkeit der Referenzdatenbanken ist auch deren Qualität
wesentlich, da z. B. Fehlbestimmungen von Arten bei der Erstellung einer solchen Datenbank zu fehlerhaften Artendetektionen bei
Metabarcoding-Erhebungen führen, die diese Referenzdatenbank nutzen.
3 Digitale Sequenzinformationen als wichtige Grundlage für die Biodiversitätsforschung
DNA-Sequenzen stellen nicht nur die grundlegenden Rohdaten jeder eDNA-Metabarcoding-Analyse dar, sondern werden hierfür auch als
eine wertvolle Ressource in Form von Referenzsequenzen benötigt. Aber auch abseits von eDNA und Metabarcoding, wie etwa in
phylogeographischen oder populationsgenetischen Untersuchungen, stellen DNA-, RNA- oder Proteinsequenzen (oftmals als DSI
zusammengefasst) eine wichtige Datengrundlage für Entscheidungen im Umweltschutz und Biodiversitätsmanagement dar. So kann man bspw.
anhand solcher Sequenzdaten feststellen, ob es bestimmte intraspezifische Strukturierungen gibt (wie Unterarten oder isolierte
Populationen), die ein separates Management erfordern. Es zeigte sich z. B., dass Bachforellen (Salmo trutta) in Österreich
südlich der Alpen zwei genetischen Hauptlinien zugehörig sind: Während ansässige, autochthone Fische der so genannten danubischen
(donaustämmigen) genetischen Linie angehören, können Fische aus Zuchten großteils der atlantischen Linie zugeordnet werden, die
natürlicherweise eher in Nordeuropa vorkommt. Rein danubischen Populationen ist daher ein besonderer Schutz zuzusprechen ( Lerceteau-Köhler et al. 2013).
Das DNA-Barcoding als ein weiterer Ansatz zielt darauf ab, bestimmte genetische Marker, auch DNA-Barcodes genannt, in den
einzelnen Arten genetisch zu charakterisieren, so dass die Arten anhand dieser Marker genetisch identifiziert werden können ( Hebert et al. 2003), was in vielen Fällen auch die Grundlage der Referenzdatenbanken für das
eDNA-Metabarcoding bildet. Hierfür gibt es mit International Barcode of Life (iBOL) eine umfassende weltweite Initiative, die zum Ziel
hat, die gesamte vielzellige Organismenwelt anhand von Barcodes zu charakterisieren. Zu diesem Zweck wurde auch eine öffentlich
zugängliche Sequenzdatenbank eingerichtet, das Barcode of Life Data System (BOLD; Hebert,
Ratnasingham 2007), in dem Forscherinnen und Forscher die generierten Sequenzdaten mit Zusatzinformationen wie
Probenursprung und Datenqualität deponieren können. Diese Sequenzinformationen stehen der Öffentlichkeit für Forschungsfragen zur
Verfügung.
Eine noch umfassendere öffentlich zur Verfügung stehende Sequenzdatenbank ist die der International Nucleotide Sequence Database
Collaboration (INSDC; https://www.insdc.org/), eines Gemeinschaftsprojekts von
Sequenzdatenbanken Europas (European Molecular Biology Laboratory's European Bioinformatics Institute – EMBL-EBI; https://www.ebi.ac.uk/), Japans (DNA Databank of Japan – DDBJ; https://www.ddbj.nig.ac.jp/) und der USA (National Center for Biotechnology
Information – NCBI, oftmals gemeinhin als GenBank bezeichnet; https://www.ncbi.nlm.nih.gov/genbank/; Benson et al. 2009). In den
individuellen INSDC-Datenbanken eingereichte Daten werden zwischen diesen automatisch ausgetauscht. Die INSDC-Datenbanken beinhalten
ein sehr breites Spektrum von DNA-Sequenzen verschiedenster genetischer Marker vielzelliger, aber auch einzelliger Organismen. Ebenso
können hier genomweite Sequenzdaten von HTS veröffentlicht werden. Andere öffentlich zugängliche Sequenzdatenbanken haben
sich, ähnlich wie BOLD, auf einen bestimmten genetischen Marker und/oder eine Organismengruppe spezialisiert, bspw. SILVA (https://www.arb-silva.de) auf ribosomale RNA-Sequenzen (rRNA) aller Organismen,
Protist Ribosomal Reference (PR2; https://pr2-database.org) auf
18S-rRNA-Sequenzen von Protisten oder UNITE (https://unite.ut.ee) auf die so genannten
ribosomalen Internal-transcribed-spacer(ITS)-Regionen von Pilzen, aber auch anderen Eukaryoten.
Sowohl BOLD als auch die INSDC-Datenbanken haben hierbei in den letzten Jahrzehnten einen rasanten Datenzuwachs erfahren – in
GenBank vor allem jene Archive, die Sequenzinformationen gesamter Genome enthalten (
Abb. 3). Dies liegt u. a. daran, dass bei der Veröffentlichung wissenschaftlicher Beiträge, die auf neu generierten
Sequenzdaten beruhen, die Autoren verpflichtet sind, diese in einer öffentlich zugänglichen INSDC-Datenbank zu deponieren.
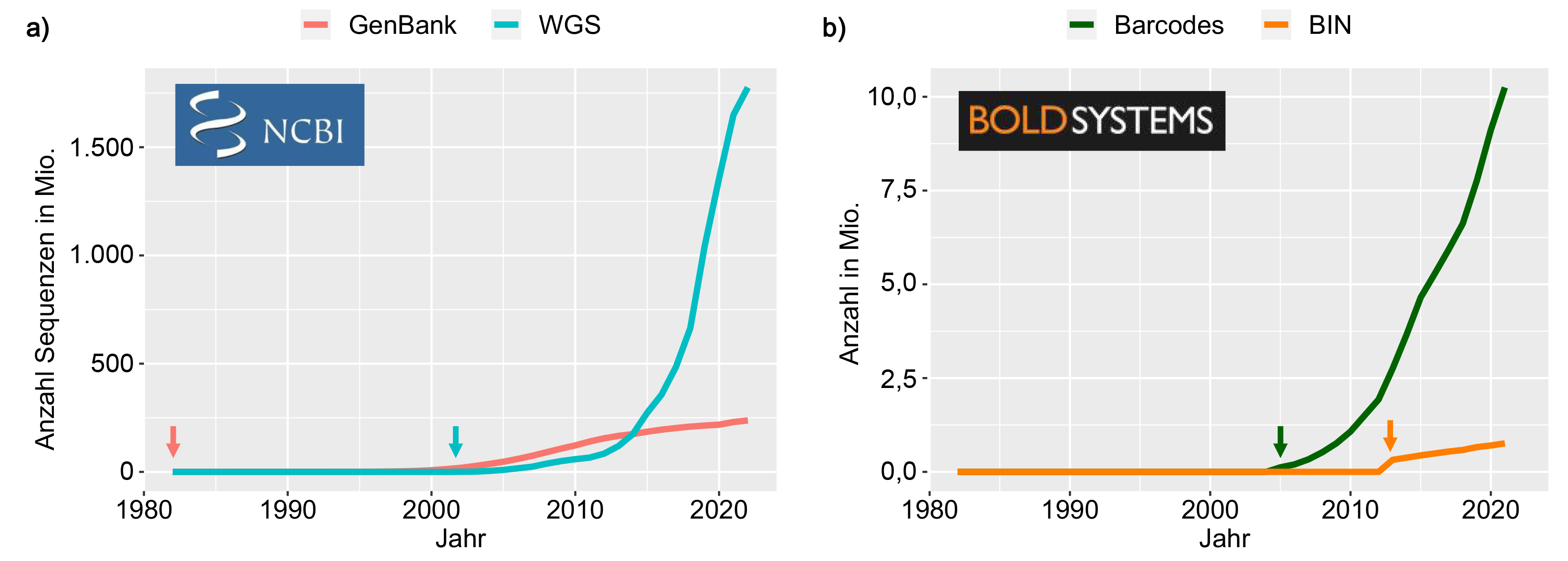
Abb. 3: Entwicklung der Datenmengen digitaler Sequenzinformationen (DSI) in zwei öffentlich zugänglichen Datenbanken –
GenBank als Beispiel der International-Nucleotide-Sequence-Database-Collaboration(INSDC)-Datenbanken und des Barcode of
Life Data System (BOLD) seit der Veröffentlichung von GenBank im Jahr 1982. a) GenBank: Anzahl der DNA-Sequenzen in der
Whole-genome-shotgun(WGS)-Datenbank der National-Center-for-Biotechnology-Information(NCBI)-GenBank (hellblau) sowie
DNA-Sequenzen von GenBank ohne die WGS-Datenbank (rosa). Die WGS-Datenbank enthält vor allem Daten aus
Gesamtgenomsequenzierungen. b) BOLD: Anzahl der Barcode-DNA-Sequenzen (grün) und der Barcode Index Numbers (BIN, orange),
die als Stellvertreter für Arten in BOLD verwendet werden. Die Pfeile zeigen das Jahr der Veröffentlichung der jeweiligen
Datenbank an (Quellen: NCBI 2022 und Ratnasingham, persönliche
Kommunikation). Fig. 3: Development of digital sequence information (DSI) data volumes in two publicly available databases – GenBank
(being a member of the INSDC collaboration) and Barcode of Life Data System (BOLD) over time since the publication of GenBank
in 1982. a) GenBank: Number of DNA sequences in the Whole Genome Shotgun (WGS) database of NCBI Genbank (cyan) and DNA
sequences from GenBank excluding the WGS database (pink). The WGS database mainly contains data from whole genome sequencing
approaches. b) BOLD: Number of DNA barcode sequences (green) and number of barcode index numbers (BINs, orange) which are used
as proxies for species in BOLD. Arrows indicate initial release of the respective database (sources:
NCBI 2022 and Ratnasingham, personal communication).
Die digitale Verfügbarkeit von Sequenzdaten hat der Biodiversitätsforschung insgesamt gewaltige Fortschritte ermöglicht. Sie
erlaubt Dateninteroperabilität auf höchster Ebene, was die Effizienz, Reproduzierbarkeit und Transparenz vieler Forschungsfelder
immens verbessert hat ( Shanmughavel 2007). So erlaubt der öffentliche Zugang zu den
Datenbanken Wissenschaftlerinnen und Wissenschaftlern auf der ganzen Welt, Sequenzdaten von Arten oder Regionen, die mitunter nur
durch teure Forschungsreisen oder Laborprozesse erzeugt werden können, zu nutzen.
Diese rasanten Entwicklungen bringen natürlich auch Herausforderungen mit sich, wie bspw. die Einrichtung von Kapazitäten, die zur
Speicherung der Daten in öffentlichen Datenbanken benötigt werden. Es müssen kontinuierlich neue Infrastrukturen aufgebaut werden, um
mit der Menge der produzierten Daten mithalten zu können ( Kodama et al. 2012). Die
Öffentlichkeit der Datenbanken birgt außerdem eine gewisse Anfälligkeit für Fehler. Die schiere Datenmenge, die täglich in GenBank
hochgeladen wird, erlaubt kaum eine Qualitätskontrolle, weshalb sich auch fehlerhafte Datensätze, z. B. durch Fehlbestimmung von Arten
oder durch Sequenzierfehler, bei GenBank, aber auch bei BOLD (das jedoch grundsätzlich striktere Qualitätskriterien besitzt) finden
( Meiklejohn et al. 2019).
Die Auswertung von Rohsequenzdaten (auch Bioinformatik genannt; Abb. 2f) ist oftmals
so komplex, dass sie spezialisiertes Personal erfordert, das eine fundierte Ausbildung benötigt. So ist es für Forscherinnen und
Forscher heutzutage kaum noch möglich, zugleich in den ökologischen/organismischen Aspekten, den Laborprozessen der Probenbearbeitung
und auch in der bioinformatischen Auswertung versiert zu sein. Durch die damit einhergehende Entkoppelung von organismischem
Fachwissen und der Kompetenz zur Auswertung der digitalen Sequenzdaten ist die enge Zusammenarbeit von Expertinnen und Experten dieser
einzelnen Teilbereiche wichtiger denn je.
4 Der Zwiespalt um den offenen Zugang zu digitalen Sequenzinformationen
Die Evolution von DSI zu einer wertvollen Ressource für Forschung und Naturschutz hat eine Diskussion über die faire Nutzung von
DSI als genetische Ressource angestoßen. Hierbei wird auf dem Übereinkommen über die biologische Vielfalt (Convention on Biological
Diversity, CBD) von 1993 aufgebaut, das neben dem Schutz und der nachhaltigen Nutzung der biologischen Vielfalt auch den gerechten
Zugang zu biologischen Ressourcen und einen entsprechenden Vorteilsausgleich zum Ziel hat. Ein Zusatzdokument, das Nagoya-Protokoll,
regelt auf bilateraler Ebene zwischen Nationen den Zugang zu genetischen Ressourcen und die gerechte Aufteilung der sich aus deren
Nutzung ergebenden Vorteile. So haben u. a. die jeweiligen Ursprungsnationen grundsätzlich das Hoheitsrecht über ihre genetischen
Ressourcen. Die Regelung bezüglich DSI, die auf den genetischen Ressourcen eines Landes beruhen, ist allerdings noch unklar. Den
freien Zugang zu DSI empfinden einige Länder mit hoher Biodiversität, oftmals Entwicklungs- oder Schwellenländer, als Untergrabung
ihrer Hoheitsrechte bezüglich ihrer genetischen Ressourcen ( Scholz et al.
2022).
Allerdings wird der freie Zugang der Wissenschaft zu DSI als eine notwendige Voraussetzung zur Erfüllung der Ziele des Global
Biodiversity Framework (ein Nachfolgeplan der CBD zum Schutz der Biodiversität zwischen 2020 und 2030) und der Ziele für nachhaltige
Entwicklung (Sustainable Development Goals – SDG) der Vereinten Nationen gesehen ( IPBES
2019). Eine Limitierung des Zugangs zu DSI würde dazu führen, dass bspw. Forschungsgruppen unterschiedlicher Länder
dieselben Daten mehrfach unabhängig voneinander produzieren müssten. Dies würde die Geschwindigkeit und Kosteneffizienz vieler
Forschungs- und Naturschutzvorhaben drastisch reduzieren, wenn nicht sogar die Vorhaben unmöglich machen, wenn bspw. die betreffenden
Einrichtungen nicht die finanziellen Mittel zur selbstständigen Generierung der Daten besitzen oder die Sammlung des genetischen
Materials aus Genehmigungs- oder Kostengründen selbst nicht durchführen können. Auch internationale Kollaborationen würden vermutlich
stark beeinträchtigt werden, da die Mitnutzung von DSI durch internationale Partner einer nationalen Forschungsgruppe verkompliziert
würde.
Die Zwickmühle im Umgang mit DSI und der wachsende Druck durch die Biodiversitätskrise sowie das soziale und wirtschaftliche
globale Ungleichgewicht verlangen nach Lösungen. Forscherinnen und Forscher sehen hierfür den Weg nach vorne weniger in einer
bilateralen Lösung wie dem Nagoya-Protokoll, sondern in einem multilateralen Ansatz, der den offenen Zugang zu DSI vom
Vorteilsausgleich entkoppelt, Bürokratie der Prozesse reduziert, Biodiversitätsforschung fördert und alle am Prozess Beteiligten fair
behandelt ( Brink, van Hintum 2022; DSI
2022; Scholz et al. 2022). Die diesbezügliche Diskussion dauert noch an,
wobei zu wünschen ist, dass es zu einer raschen und allseits zufriedenstellenden Lösung kommt, damit DSI der Forschung, dem
Naturschutz, aber auch der globalen Gesellschaft gleichermaßen zugutekommen können.
5 Literatur
↑
Barnes M.A., Turner C.R. (2016): The ecology of environmental DNA and implications for conservation genetics.
Conservation Genetics 17: 1 – 17.
↑
Belle C.C., Stoeckle B.C., Geist J. (2019): Taxonomic and geographical representation of freshwater environmental DNA research in aquatic
conservation. Aquatic Conservation: Marine and Freshwater Ecosystems 29: 1.996 – 2.009.
↑
Beng K.C., Corlett R.T. (2020): Applications of environmental DNA (eDNA) in ecology and conservation: Opportunities, challenges and prospects.
Biodiversity and Conservation 29: 2.089 – 2.121.
↑
Benson D.A., Karsch-Mizrachi I. et al. (2009): GenBank. Nucleic Acids Research 37: D26 – D31.
↑
Bohmann K., Evans A. et al. (2014): Environmental DNA for wildlife biology and biodiversity monitoring. Trends in Ecology and Evolution 29:
358 – 367.
↑
Brink M., van Hintum T. (2022):
Practical consequences of digital sequence information (DSI) definitions and access and benefit-sharing scenarios from a plant
genebank's perspective. Plants, People, Planet 4: 23 – 32.
↑
Buchner D., Macher T.-H. et al. (2021): Standardized high-throughput biomonitoring using DNA metabarcoding: Strategies for the adoption of automated
liquid handlers. Environmental Science and Ecotechnology 8: e100122. DOI: 10.1016/j.ese.2021.100122
↑
Clare E.L., Economou C.K. et al. (2022): Measuring biodiversity from DNA in the air. Current Biology 32: 693 – 700.
↑
Darling J.A., Galil B.S. et al. (2017): Recommendations for developing and applying genetic tools to assess and manage biological invasions in marine
ecosystems. Marine Policy 85: 54 – 64.
↑
Deiner K., Bik H.M. et al. (2017):
Environmental DNA metabarcoding: Transforming how we survey animal and plant communities. Molecular Ecology 26:
5.872 – 5.895.
↑
DSI/Digital Sequence Information (2022): Open Letter. https://www.dsiscientificnetwork.org/open-letter/
(aufgerufen am 28.6.2022).
↑
Dussling U. (2009): Handbuch zu fiBS. Schriftenreihe des Verbandes Deutscher Fischereiverwaltungsbeamter und
Fischereiwissenschaftler e. V., Heft 15: 41 S.
↑
Gloor G.B., Macklaim J.M. et al. (2017): Microbiome datasets are compositional: And this is not optional. Frontiers in Microbiology 8: e02224. DOI:
10.3389/fmicb.2017.02224
↑
Glover-Kapfer P., Soto-Navarro C.A., Wearn O.R. (2019): Camera-trapping version 3.0: Current constraints and
future priorities for development. Remote Sensing in Ecology and Conservation 5: 209 – 223.
↑
Harper L.R., Buxton A.S. et al. (2019): Prospects and challenges of environmental DNA (eDNA) monitoring in
freshwater ponds. Hydrobiologia 826: 25 – 41.
↑
Hebert P.D., Cywinska A. et al. (2003): Biological identifications through DNA barcodes. Proceedings of the Royal Society B: Biological Sciences 270:
313 – 321.
↑
Hebert P.D., Ratnasingham S. (2007): The Barcode of Life Data System BOLD. Molecular Ecology Notes 7: 355 – 364.
↑
Hering D., Borja A. et al. (2018): Implementation options for DNA-based identification into ecological status assessment under the European Water
Framework Directive. Water Research 138: 192 – 205.
↑
IPBES/Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services (2019): Global assessment report on biodiversity and
ecosystem services of the Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services. IPBES Secretariat.
Bonn: 1.148 S.
↑
Jerde C.L., Mahon A.R. et al. (2011): “Sight-unseen” detection of rare aquatic species using
environmental DNA. Conservation Letters 4: 150 – 157.
↑
Kodama Y., Shumway M., Leinonen R. (2012): The sequence read archive: Explosive growth of sequencing data. Nucleic Acids Research 40:
2.011 – 2.013.
↑
Kolter A., Gemeinholzer B. (2020): Plant DNA barcoding necessitates marker-specific efforts to establish more comprehensive reference databases.
Genome 64(3): 265 – 298.
↑
Kupfer A., Schlüpmann M. (2009): Methoden der Amphibienerfassung: ein Überblick. Zeitschrift für
Feldherpetologie 15: 7 – 84.
↑
Lerceteau-Köhler E., Schliewen U. et al. (2013): Genetic variation in brown trout Salmo trutta across the Danube, Rhine, and Elbe headwaters: A
failure of the phylogeographic paradigm? BMC Evolutionary Biology 13: 176.
↑
Lynggaard C., Bertelsen M.F. et al. (2022): Airborne environmental DNA for terrestrial vertebrate community monitoring. Current Biology 7:
701 – 707.
↑
Lyon J.P., Bird T. et al. (2014): Efficiency of electrofishing in turbid lowland rivers: Implications for measuring
temporal change in fish populations. Canadian Journal of Fisheries and Aquatic Sciences 71: 878 – 886.
↑
Macher T.-H., Schütz R. et al. (2021): Beyond fish eDNA metabarcoding: Field replicates disproportionately improve the detection of stream associated
vertebrate species. Metabarcoding and Metagenomics 5: e66557.
↑
Meier C., Haase P. et al. (2006): Methodisches Handbuch Fließgewässerbewertung – Handbuch zur
Untersuchung und Bewertung von Fließgewässern auf der Basis des Makrozoobenthos vor dem Hintergrund der EG-Wasserrahmenrichtlinie.
https://gewaesser-bewertung.de/index.php?article_id=124 (aufgerufen am 29.6.2022).
↑
Meiklejohn K.A., Damaso N. et al. (2019): Assessment of BOLD and GenBank – Their accuracy and reliability for the
identification of biological materials. PLOS ONE 14: e0217084.
↑
Miya M., Sato Y. et al. (2015): MiFish, a set of universal PCR primers for metabarcoding environmental DNA from fishes: Detection of more than
230 subtropical marine species. Royal Society Open Science 2(7): e150088.
↑
NCBI/National Center for Biotechnology Information (2022): GenBank and WGS Statistics. https://www.ncbi.nlm.nih.gov/genbank/statistics (aufgerufen am 8.6.2022).
↑
Pawlowski J., Apothéloz-Perret-Gentil L., Altermatt F. (2020): Environmental DNA: What's behind the term? Clarifying the terminology
and recommendations for its future use in biomonitoring. Molecular Ecology 29: 4.258 – 4.264.
↑
Pawlowski J., Bonin A. et al. (2021): Environmental DNA for biomonitoring. Molecular Ecology 30:
2.931 – 2.936.
↑
Pawlowski J., Kelly-Quinn M. et al. (2018): The future of biotic indices in the ecogenomic era:
Integrating (e)DNA metabarcoding in biological assessment of aquatic ecosystems. Science of the Total Environment 637 – 638:
1.295 – 1.310.
↑
Pimm S.L., Jenkins C.N. et al. (2014): The biodiversity of species and their rates of extinction, distribution, and protection. Science 344(6187):
e1246752.
↑
Rees H.C., Maddison B.C. et al. (2014): REVIEW: The detection of aquatic animal species using environmental
DNA – A review of eDNA as a survey tool in ecology. Journal of Applied Ecology 51: 1.450 – 1.459.
↑
Rodgers T.W., Xu C.C. et al. (2017): Carrion fly-derived DNA metabarcoding is an effective tool for mammal surveys: Evidence from a known tropical
mammal community. Molecular Ecology Resources 17(6): e133 – e145.
↑
Rodriguez-Ezpeleta N., Morissette O. et al. (2021): Trade-offs between reducing complex terminology and producing accurate interpretations from
environmental DNA: Comment on “Environmental DNA: What's behind the term?” by Pawlowski et al., (2020). Molecular Ecology 30:
4.601 – 4.605.
↑
Ruppert K.M., Kline R.J. et al. (2019): Past, present, and future perspectives of environmental DNA (eDNA) metabarcoding: A
systematic review in methods, monitoring, and applications of global eDNA. Global Ecology and Conservation 17:
1 – 29.
↑
Scholz A.H., Freitag J. et al. (2022): Multilateral benefit-sharing from digital sequence information will support both science and biodiversity
conservation. Nature Communications 13: 1.086.
↑
Shanmughavel P. (2007): An overview on biodiversity information in databases. Bioinformation 1: 367 – 369.
↑
Stat M. (2017): Ecosystem
biomonitoring with eDNA: Metabarcoding across the tree of life in a tropical marine environment. Scientific Reports 7:
e12240.
↑
Südbeck P. (2005): Methodenstandards zur Erfassung der Brutvögel Deutschlands. Dachverband
Deutscher Avifaunisten. Steckby: 792 S.
↑
Taberlet P., Coissac E. et al. (2012): Environmental DNA. Molecular Ecology 21: 1.789 – 1.793.
↑
Van Dijk E.L., Auger H. et al. (2014): Ten years of next-generation sequencing technology. Trends in Genetics 30: 418 – 426.
↑
Vörös J., Márton O. et al. (2017): Surveying Europe's only cave-dwelling Chordate species (Proteus anguinus)
using environmental DNA. PLOS ONE 12: e0170945.
↑
Weigand H., Beermann A.J. et al. (2019): DNA barcode reference libraries for the monitoring of aquatic biota in Europe: Gap-analysis and recommendations
for future work. Science of the Total Environment 678: 499 – 524.
Förderung
Dieser Artikel wurde ermöglicht durch den Austrian Science Fund (FWF): 35059-B.
Fußnoten

